Table of Contents
IgA Nephropathy may be the most common glomerulonephritis (GN) in the world. It is most common in East Asians (Chinese and Japanese), relatively common in Caucasians and relatively rare in people of African descent.
IgA nephropathy can have a wide range of presentations with variable prognosis.
This article will discuss:
- Clinical presentations
- Prognosis (assessment of risk of progressive CKD)
- Clinical Features
- Biopsy Features
- Treatment
It all comes down to this:
- Proteinuria is bad: More proteinuria = worse prognosis
- Scarring is bad: More scarring on biopsy or lower baseline eGFR (presumed more scarring) = worse prognosis
- Inflammation (on biopsy): Suggests potential response to immunosuppressive treatment
You can stop reading here and know more than most. Keep reading to know a lot more than most.
Clinical Presentations
Clinical Presentations: More Details
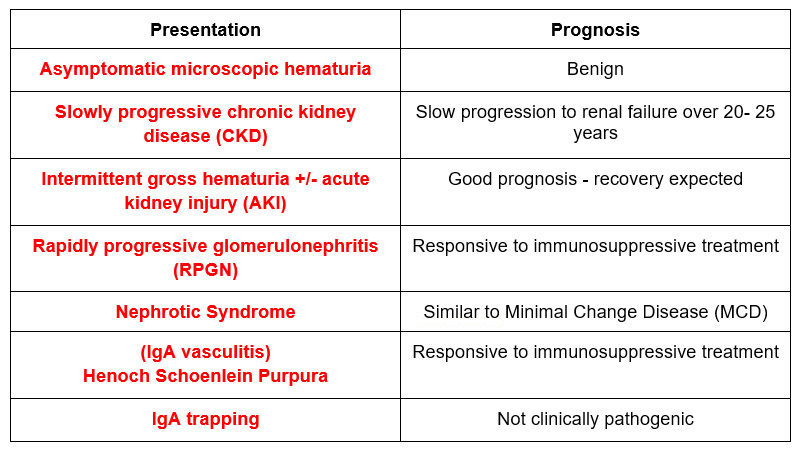
Asymptomatic microscopic hematuria
Patients may present with this and little to no proteinuria with normal kidney function. This has a good (benign) prognosis. The differential includes:
- GU causes of hematuria
- Thin basement membrane disease (benign familial hematuria):
- Alport’s syndrome
Slowly progressive CKD
These patients typically have proteinuria, with or without microscopic hematuria and/or decreased eGFR. 25-30% of patients have this presentation with a slow progression to kidney failure over 20-25 years.
Intermittent gross hematuria
This occurs in the context of an infection (an upper respiratory or gastrointestinal infection). These cases can present with AKI, even with crescents on kidney biopsy. But they get better. Recovery should occur within 1 week, if not then biopsy. Biopsy may show limited crescents (<25% of glomeruli). The differential includes:
- Post infectious (post streptococcal) GN. The difference: In IgA nephropathy; the hematuria occurs at the time of the infection; it is referred to as synpharyngitic. In post infectious / post streptococcal hematuria will occur 1-2 weeks after the infection.
- Membranoproliferative GN (MPGN). The difference: in MPGN hematuria (or microhematuria) will persist chronically
Post-infectious GN and MPGN are associated with low complements (C3 in post-infectious C3 and C4 in MPGN). IgA typically has normal serum complements.
Rapidly progressive glomerulonephritis
This will be associated with a quicker decline in kidney function (>50% decrease in eGFR over < 3 months). Biopsy will show signs of active inflammation such as cellular crescents and/or endocapillary inflammation. It may be difficult to distinguish from the Intermittent gross hematuria presentation above. If there is > 50% glomeruli with crescents treatment is indicated, if < 50% crescents (especially if kidney function is not worsening) then don’t.
Nephrotic syndrome
If it presents like Minimal Change Disease and looks like MCD on light microscopy, then treat it as such.
IgA vasculitis (Henoch Schoenlein purpura)
This presents with abdominal pain and vasculitic rash. There may be associated renal involvement with a nephritic picture.
IgA trapping
There are times where a kidney biopsy immunofluorescence be positive for IgA that is not pathogenic or clinically significant. This may happen in liver disease (decreased IgA clearance by Kupffer cells) or celiac disease (increased IgA production from dietary antigen).
Prognosis
Clinical Features
- Proteinuria: The worse the proteinuria the worse the prognosis. For this reason it is often recommended to use steroids to treat patients with IgA nephropathy with > 1 gram proteinuria/ day (despite RAAS blockers).
- Decreased kidney function: For this reason it is often recommended to NOT use steroids to treat patients with advanced kidney disease (ie eGFR < 30). Unless this is an acute nephritic RPGN picture.
Pathologic Features
- There is a standard classification system for IgA Nephropathy biopsies called the MEST-C This score provides prognostic information.
M – Mesangial hypercellularity
E – Endocapillary hypercellularity
S– Segmental glomerulosclerosis
T – Tubular atrophy/Interstitial fibrosis
C – Crescents
There is a prediction calculator that gives the risk of a 50% decline in eGFR or development of kidney failure at specified intervals. You can find it here: International IgAN Prediction Tool at biopsy – Adults | QxMD
Treatment
The treatment of this condition is difficult. There is not a lot of good data for the effectiveness of immunosuppressants. The key is to find what niche, what clinical context immunosuppressants may be beneficial. For the majority this is supportive care, some situations warrant steroids or cyclophosphamide.
Supportive care
- (Non specific CKD treatment). Indicated with CKD +/- proteinuria. My approach to proteinuria here:
- BP management: Target < 130/80
- Proteinuria > 500 mg/day:
- Renin angiotensin aldosterone system (RAAS) blockers (ACE-I or ARBs) at maximally tolerated doses. Do not use both together – no additional benefit found
- SGLT 2 inhibitors: Add on to RAAS blockers if proteinuria remains > 500 mg per day. A number of patients in the DAPA – CKD trial (link A pre-specified analysis of the DAPA-CKD trial demonstrates the effects of dapagliflozin on major adverse kidney events in patients with IgA nephropathy ) had IgA nephropathy
- Sparsentan (Filspari): This is a dual ARB / endothelin 1 receptor antagonist. It received conditional FDA approval based on this study: (link Sparsentan in patients with IgA nephropathy: a prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial – The Lancet ) as it reduced proteinuria more than an ARB alone.
Steroids
- If persistent proteinuria > 1 gram per day despite RAAS blockers
- Prednisone: 0.8 – 1.0 mg/kg /day for 8 weeks; taper 5-10 mg/day every 2 weeks – total duration 6-8 months
- Methylprednisolone: 0.4 mg/kg/day (max 32 mg) x 2 months; taper 4 mg/day per month x 4 months
- Targeted release budesonide (Tarpeyo): This was approved for proteinuria > 2 grams per day (or protein creatinine ratio > 1.5 gram per gram)The idea is this: This drug is released in the terminal ileum. The terminal ileum is where the peyer patches that release IgA are. Therefore this steroid formulation can be effective with fewer adverse effects. The dose is 16 mg per day for 9 months, taper by using 8 mg per day for 2 weeks.
Can also use steroids for IgA vasculitis (Henoch Schonlein Purpura)
Avoid steroids if:
- eGFR < 30
- High risk for steroid complications (ie diabetes, obesity; osteoporosis)
- Latent infections (Hepatitis B/TB; osteoporosis)
-
Cyclophosphamide:
- RPGN with crescents
- Severe IgA vasculitis (Henoch Schonlein Purpura)
-
Mycophenolate Mofetil/ Hydroxychloroquine
- Chinese patients with proteinuria.
- Some would consider MMF in other patient populations if there was an inability to take steroids. Limited studies.
-
Tonsillectomy
- Japanese patients. Apparently this is the standard of care in Japan.
Summary
IgA nephropathy is the most common glomerulonephritis in the world. It is caused by under galactosylated IgA1. There may be a genetic predisposition to form under galactosylated IgA1 with an additional environmental antigenic trigger. These abnormal IgA1 antibodies can self aggregate or other IgA or IgG antibodies against them can form immune complexes which then deposit in the kidney glomeruli.
There are a wide range of clinical presentations, many benign (asymptomatic microscopic and intermittent gross hematuria), some with slowly progressive CKD. A major risk factor for progression is the degree of proteinuria which should be treated with supportive care (BP control, RAAS blockers +/- SGLT 2 inhibitors) and immunosuppressives (most often corticosteroids) in those with the highest risk of progression.

{kind=link}
{kind=link}
{kind=link}