Table of Contents
Alport syndrome is the most common inherited kidney disease, more prevalent than autosomal dominant polycystic kidney disease (ADPKD).
Awareness of this condition, particularly the autosomal recessive and autosomal dominant variants, has increased as genetic testing has become mainstream.
What is Alport syndrome?
Alport syndrome is a genetic condition with an abnormality of type 4 collagen. Type 4 collagen is an integral part of basement membranes in the body. There are 6 alpha chains of type 4 collagen.
The genes of interest in Alport syndrome are for the alpha 3,4 and 5 chains of type 4 collagen. These form a heterotrimer that comprise basement membranes in the:
- Kidney: Glomerular basement membrane (GBM)
- Ear: Basement membrane in cochlea
- Eye: Basement membrane at base of ocular lens
The clinical manifestations of Alport syndrome therefore affect these organs.
- Kidney: Hematuria, proteinuria, CKD, ESRD
- Ear: Sensorineural hearing loss
- Eye: Anterior lenticonus, fleck retinopathy
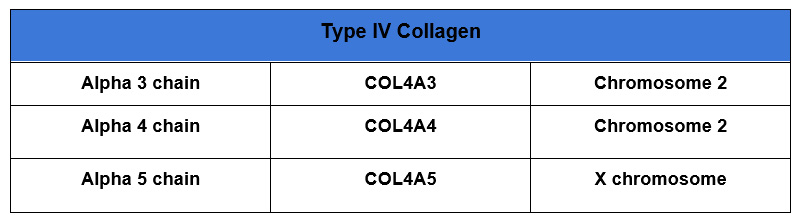
The genes for alpha 3 and alpha 4 of type 4 collagen are located close together on chromosome 2. Therefore mutations in both these genes can be co-transmitted.
Types of Alport Syndrome
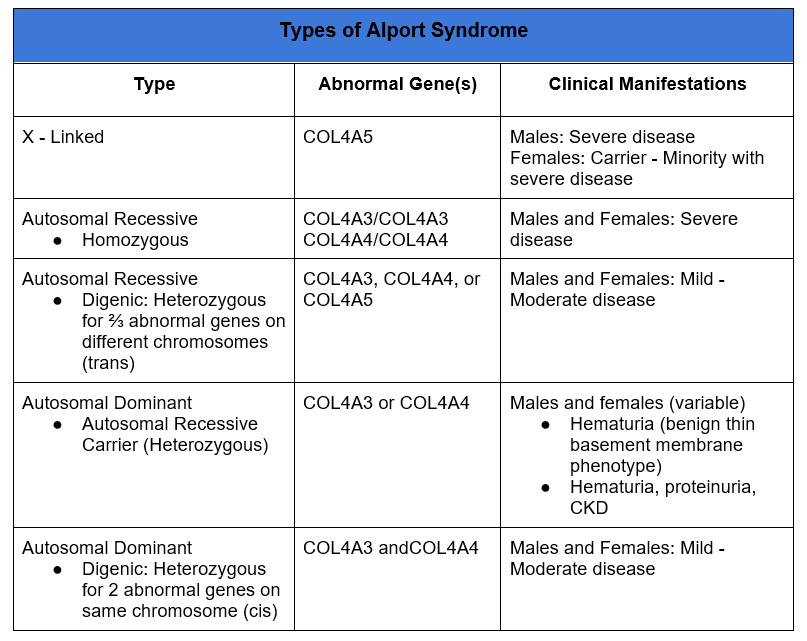
X linked
- “Traditional” Alport syndrome well known cause of ESRD
- Abnormal gene is COL4A5.
- Typically affects males. Males have one X chromosome, females have two X chromosomes so would be a carrier. Transmission from mother to son, with maternal uncles potentially affected.
- Female (carriers) may also be affected, via a phenomenon referred to as lyonization. In female cells one of the two X chromosomes is randomly inactivated during embryonic development, to prevent double expression of X-linked proteins. This results in a mosaic pattern with some cells expressing the phenotype of X linked Alport, others expressing a normal phenotype.
Clinical Manifestations
- Incidence 1: 2000
- High risk of kidney failure
- Most males
- Age 25 – 50%
- Age 40 – 90%
- Age 60 – 100%
- 15-30% Females
- Later in life
- Hematuria most males, 95% females
- Proteinuria, progressive CKD
- Sensorineural hearing loss
- Ocular abnormalities
- Anterior lenticonus
- Fleck Retinopathy
- Most males
Pathology
- Characteristic glomerular basement membrane abnormalities on electron microscopy.
- Initial thinning of the GBM
- Subsequent splitting of the basement membrane resulting in lamellated pattern (of thin layers)
Treatment
- No disease specific treatment
- RAS blockers; SGLT 2 inhibitors; blood pressure control slow progression
Autosomal Recessive
Abnormalities in COL4A3 and/or COL4A4 genes. Located in close proximity on chromosome 2.
Less Common.
- Homozygous: 2 abnormal COL4A3 or COL4A4 genes
- Compound heterozygous: 2 abnormal COL4A3 or COL4A4 genes, with different mutations
- Digenic: 2 abnormal genes (COL4A3, COL4A4 or COL4A4)
Clinical Manifestations
- Affects men and women equally
- Homozygous/compound heterozygous:
- Severe manifestations (similar to X-linked males). Most will develop kidney failure.
- Sensorineural hearing loss
- Digenic
- Mild – moderate manifestations
Autosomal Dominant
COL4A3 and COL4A5 heterozygotes, in addition to being autosomal recessive carriers, also express an autosomal dominant phenotype.
- Autosomal recessive carrier: Offspring at risk for autosomal recessive disease if other parent also carrier. (< 1% offspring will have autosomal recessive or digenic Alport syndrome).
- Autosomal Dominant: Heterozygotes have variable clinical manifestations.
- Digenic: If abnormalities for COL4A3 and COL4A4 are cis can be transmitted in autosomal dominant manner.
Awareness of this variant has increased with the more common use of genetic testing.
heterozygotes 10-20% – historically but probable less – with genetic testing
Clinical Manifestations:
- Microscopic glomerular hematuria. Most will have. Historically, referred to as thin basement membrane disease (benign familial hematuria) presented with asymptomatic hematuria, without proteinuria or decrease in GFR with biopsy findings of thin basement membranes on electron microscopy.
- Non nephrotic proteinuria (approximately ⅔)
- CKD (Approximately ⅓). CKD is relatively mild and progression to ESRD rare.
- Hearing and ocular abnormalities are rare
- Focal segmental glomerulosclerosis (FSGS).
It is recommended that COL4A3 or COL4A4 heterozygotes with autosomal dominant Alport syndrome not be kidney transplant donors.
Genetic Testing
X-linked Alport syndrome is usually clinically apparent given the characteristic clinical findings and renal biopsy findings and often positive family history.
There are panels that test for known genes associated with kidney disease. These are relatively simple and have become more mainstream. The patient receives a kit at home, performs a cheek swab which is returned via mail, similar to Ancestry and 23 and me.
Renasight – Genetic Testing for Kidney Disease
Genetic testing is recommended for:
- Family history in 1st degree family members
- Persistent glomerular, dysmorphic hematuria ( > 6 months) without known cause
- Persistent proteinuria > 500 mg day without known cause
- Focal segmental glomerulosclerosis (FSGS) especially if steroid resistant and/or familial
- Familial IgA nephropathy
- CKD of unknown etiology
For a detailed review check out this article:
Guidelines for Genetic Testing and Management of Alport Syndrome – PMC
Summary
Alport syndrome is a genetic disease with an abnormality in the alpha 3,4 or 5 chain of type 4 collagen. The X linked variant is a cause of ESRD with associated hearing loss and ocular abnormalities in males. Females are carriers, but may have manifestations of hematuria and kidney disease due to mosaicism. Autosomal recessive and dominant forms typically involve abnormalities of alpha 3 or alpha 4 chains and affect men and women equally. The homozygous autosomal recessive form is more severe with associated kidney failure and hearing loss. The heterozygous form is both autosomal dominant and a carrier for autosomal recessive. It is associated with hematuria. A lesser percentage will develop proteinuria and CKD.

{kind=link}
{kind=link}
{kind=link}